Research Summary
Characterization of oncogenic mechanisms NOTCH1 in T-ALL
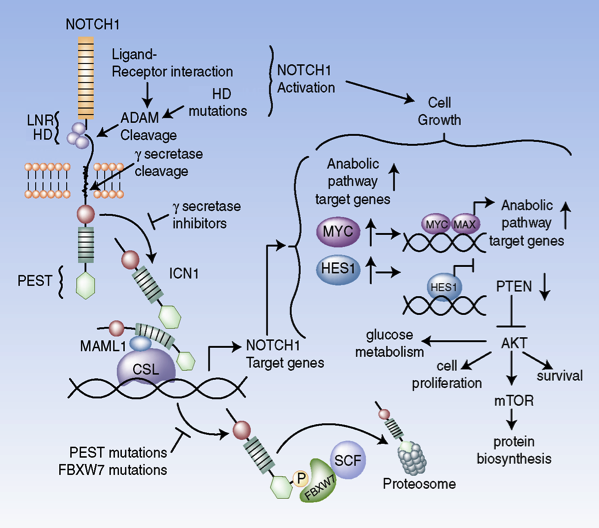
Figure 1. Oncogenic pathways controlled by NOTCH1 signaling in T-ALL (Palomero et al. PNAS 2006; Palomero et al. Nat Med 2007; Margolin et al. PNAS 2009; Herranz et al. Nat Med 2014; Herranz et al. Nat Med 2015).
T-ALL is an aggressive and high risk hematologic malignancy requiring treatment with highly aggressive chemotherapy. The introduction of intensified therapies has improved clinical outcomes in this disease, however 20% of children and 50% of adults with T-ALL still relapse and die of refractory leukemia. In this context, our NOTCH1 research program seeks to identify targetable oncogenic mechanisms for the development of new targeted anti-leukemic drugs.
NOTCH1 was first proposed as pathogenic factor in T-ALL from the study of the translocation t(7; 9) (q34; q34.3), which induces expression of a truncated and constitutively active form NOTCH1 in 1% of T-ALL cases. However, the central role of NOTCH1 in this disease was only realized with identification of activating mutations in the NOTCH1 gene present in over 60% of T-ALL patients 1. The fundamental importance of these mutations in T-ALL is underlined by the potential of NOTCH1 as a therapeutic target, as small molecule inhibitors of the gamma-secretase complex, an aspartyl protease strictly necessary for the activity of NOTCH1, can effectively block the oncogenic activity of NOTCH1 in T-ALL. Over the last years our research program has addressed the most important issues with translational impact for the clinical development of anti-NOTCH1 therapies focusing on the molecular mechanisms that mediate the oncogenic activity of NOTCH1 and the effects of NOTCH1 inhibition in T-ALL (Figure 1). To this end we made the first systematic characterization of transcriptional programs and direct targets regulated by NOTCH1 in T-ALL identifying MYC as a critical factor in T-cell transformation by activated NOTCH1 2,3. Characterization of NOTCH1 DNA binding sites across the genome showed that transcriptional activation of MYC in these leukemias is controlled through N-Me, a NOTCH1-dependent enhancer located 1.4Mb from the MYC promoter. The central role of N-Me both in the development of T-cell precursors and in the context of T-ALL is demonstrated by the loss of expression of Myc and defects in thymocyte development and the oncogenic activity of NOTCH1 in conditional knockout mice deficient for this enhancer 4. Beyond these animal models, the importance of N-Me in human leukemias is highlighted by the identification of recurrent and focal chromosomal duplications centered on this enhancer in T-ALL.
Despite the central role of NOTCH1 in the pathogenesis of T-ALL, gamma-secretase inhibitors have shown limited therapeutic activity in the clinic highlighting the importance of elucidating mechanisms of resistance and the identification of new therapeutic combinations that would be able to increase their antitumor activity of anti-NOTCH1 therapies. Through analysis of tumors that are sensitive or resistant to the action of gamma-secretase inhibitors we first identified the mutational loss of PTEN, a tumor suppressor gene involved in the negative regulation of the PI3K pathway, in 20% of human T-ALLs and established a direct correlation between PTEN loss and resistance to NOTCH1-suppression therapies 5. Moreover, we verified that that Pten loss and consequent activation of PI3K signaling in a mouse model of NOTCH1-induced leukemia induce resistance to gamma-secretase inhibitors in vivo 6. Furthermore, and using metabolomics and transcriptomic profiling we demonstrated that resistance to NOTCH1 inhibition is a result of Pten loss and is mediated by metabolic reprograming characterized by increased glycolysis and suppression of autophagy. Finally, it is important to note that these studies also identified glutaminolysis inhibition and suppression of autophagy as new therapeutic targets synergistic with NOTCH1 inhibition in the treatment of T-ALL.
Genetic analysis of resistance to chemotherapy in acute lymphoblastic leukemia
Movie 1. NT5C2 Mutations Drive Resistance to 6-MP Chemotherapy.
Relapse and resistance to therapy are the most important challenges in the treatment of ALL. Based on the hypothesis that cells of acute lymphoblastic leukemia are genetically heterogeneous we proposed that the cytotoxic effects of chemotherapy may lead to the selection of specific mutations involved in resistance to therapy. To identify these alterations and the evolutionary history of leukemias during disease progression and relapse we performed the first global mutational analysis of matched diagnosis and relapse ALL samples (Figure 2). Clonal evolution analyses revealed that the leukemic clones responsible for the progression and relapse originate from ancestral populations that are genetically related to, but distinct from the dominant primary clone present at diagnosis, through a pattern of branched evolution. This means that genetic selection in the initial stages of the disease (before treatment) may favor the selection of dominant leukemic populations in terms of growth and proliferation, while subclonal populations that are more latent, but at the same time more resistant to therapy, are selected during treatment and are the ones that ultimately contribute to relapse. In addition, and most notably, analysis of relapse associated mutations in the context of this study identified the presence of recurrent mutations in NT5C2 as a highly prevalent event in relapsed ALL18.
NT5C2 encodes for a nucleotidase responsible for the intracellular clearance of 6-mercaptopurine, a nucleoside analogue with cytotoxic activity used in the treatment of ALL. Relapse-associated NT5C2 mutations are gain of function, result in increased nucleotidase activity and induce resistance to 6-MP in cellular assays. Consistently, NT5C2-mutant tumors show early relapse and progression under 6-MP treatment in the clinic. These results have profound implications for the clinical management of relapse ALL, support the need to implement new monitoring strategies aimed at detecting subclonal resistance-associated mutations and identify NT5C2 as a potential therapeutic target for the treatment of relapsed leukemias with resistance to 6-MP.
Analysis of the mechanisms of glucocorticoid resistance in T-ALL
Identifying drugs that synergistically improve anti-leukemic effects of anti-NOTCH1 therapies is critical to the development of new effective regimens for the treatment of T-ALL. Glucocorticoids play a key role in the treatment of ALL and glucocorticoid resistance is associated with poor prognosis in the clinic. In this context, and considering that the effects of NOTCH1 in promoting cell growth oppose the pro-apoptotic effects of glucocorticoids in T-cell progenitor cells, we first proposed that NOTCH1 might contribute to glucocorticoid resistance in T-ALL. Following this hypothesis we demonstrated that inhibition of NOTCH1 signaling effectively reverses glucocorticoid resistance in cell lines and primary human leukemias 7. Mechanistically, we showed that HES1, a transcriptional repressor controlled by NOTCH1 suppresses a self-regulating positive feedback loop controlling glucocorticoid receptor auto-upregulation that is critical for induction of apoptosis. Notably, the combination of gamma secretase inhibitors and glucocorticoids is not only effective against glucocorticoid resistant leukemias, but also suppresses the intestinal toxicity typically associated with systemic NOTCH inhibition. The enteroprotective effects of glucocorticoids in the context of gamma secretase inhibitor therapy is the result of a previously unrecognized effect of corticoids in antagonizing the goblet cell metaplasia induced by Notch inhibition. These results support that therapies combining NOTCH1 inhibition and glucocorticoids can have enhanced anti-leukemic activity with low toxicity and are the basis of new clinical trials currently testing the therapeutic efficacy of this treatment combination in patients with refractory T-ALL.
In addition, and using reverse engineering of signaling and transcriptional networks we identified a direct role of the AKT1 kinase, a central factor in the PI3K signaling pathway, as a major modulator of glucocorticoid response in T-ALL. Following on these results we demonstrated that AKT1 directly phosphorylates the glucocorticoid receptor and limits its transcriptional activity. Accordingly, activation of PI3K-AKT induced by PTEN loss causes resistance to steroid treatment, whereas pharmacological inhibition of AKT reverses this phenotype and augments the anti-leukemic activity of corticosteroids. These results demonstrate the central role of the PI3K-AKT signaling pathway as an inducer of glucocorticoid resistance in T-ALL and support combination therapy with inhibitors of PI3K-AKT and glucocorticoids in this disease 8.
Functional characterization of the TLX1 and TLX3 oncogenes T-ALL
The presence of aneuploidy is a highly prevalent phenomenon in human leukemias. However the mechanisms that lead to the gain and loss of chromosomes during leukemic transformation are poorly understood. TLX1 and TLX3 are two oncogenic transcription factors activated by chromosomal translocations in one third of T-ALL cases. In order to characterize the oncogenic role of TLX1 we generated the first transgenic mouse model of T-ALL induced by this oncogene.
Using this model we defined the transcriptional programs controlled by TLX1 and established a direct role of this oncogene as driver of aneuploidy through the abolition of mitotic checkpoints. In addition, genomic profiling of TLX1 induced leukemias identified BCL11B as a new tumor suppressor gene controlled by TLX1 and mutated in 10% of patients with T-ALL 9. Furthermore, the characterization of transcriptional networks controlled by TLX1 and TLX3 in human leukemias demonstrated an overlap in their direct targets and expression programs. These studies defined TLX1 and TLX3 as transcriptional repressors; established the full repertoire of their direct target genes. Finally, transcription network analyses identified RUNX1 as a central node in the oncogenic program of leukemias induced by TLX1 and TLX3 and predicted a tumor suppressor role for this transcription factor in T-ALL; hypothesis that was validated with the identification of RUNX1 mutations in 5% of T-ALL cases10.
Identifying new oncogenes and tumor suppressors in T-cell leukemias and lymphomas
In recent years, the Ferrando lab has pioneered the use of genomic approaches to identifying new oncogenes and tumor suppressors involved in the pathogenesis of T-ALL. One of the most enigmatic features of T-ALL is the higher incidence of this disease in males compared with females (3:1 ratio). In order to investigate a potential genetic basis for this difference, we analyzed the full exonic sequence of chromosome X in male patients with T-ALL. These studies identified the presence of mutations in the gene PHF6 in 20% of T-ALL 11. PHF6 encodes an epigenetic regulator never implicated in tumor development before. Notably, mutations in PHF6 are present almost exclusively in male patients with T-ALL. Apart from the recurrent mutations in PTEN, BCL11B and RUNX1 mentioned before, we identified SH2B3 as a tumor suppressor with germline and somatic mutations in ALL 12 as well as the presence of highly prevalent T-ALL mutations in WT1 13, ETV6 14, and the epigenetic factors SUZ12 and EZH2 15. Recently, we have expanded our genomic analyses to the study of Peripheral T-cell Lymphomas (PTCLs), a group of hematological malignancies associated with poor prognosis. Thus, combining sequence analysis of exomes and RNAseq data we identified a new highly prevalent mutation in RHOA (RHOA G17V), present in nearly 70% of cases of angioimmunoblastic T-cell lymphoma in conjunction with epigenetic mutations in TET2, IDH2 and DNMT3A 16. These studies also uncovered activating mutations in FYN tyrosine kinase gene sensitive to dasatinib in PTCL not otherwise specified tumors and defined the overall mutational landscape and a critical pathogenic role of TCR-signaling in aggressive forms of cutaneous T-cell lymphoma 17. In all, these studies have transformed our understanding of the genetic basis of T-ALL and PTCL; established new diagnostic molecular markers; and identified new therapeutic targets with direct clinical application.
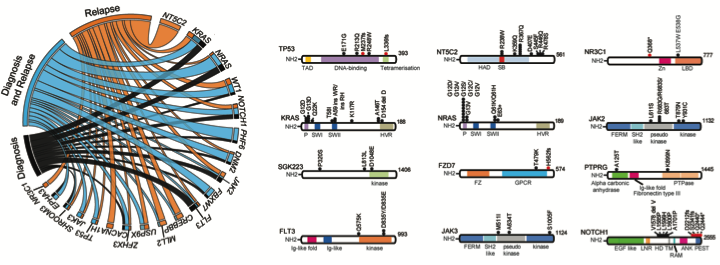
Figure 2. Exome sequencing mutation analysis of diagnosis and relapsed ALL samples. (Tzoneva Nat Med 2013)